Rate prediction for homogeneous nucleation of methane hydrate
23 September 2020
Mixtures of methane gas and water can spontaneously form a solid hydrate. Such methane hydrates naturally occur in abundance at the ocean floors and in permafrost, exceeding the natural gas reserve substantially. As such, methane hydrates are envisioned not only as a future energy resource but also as very relevant for global climate change.
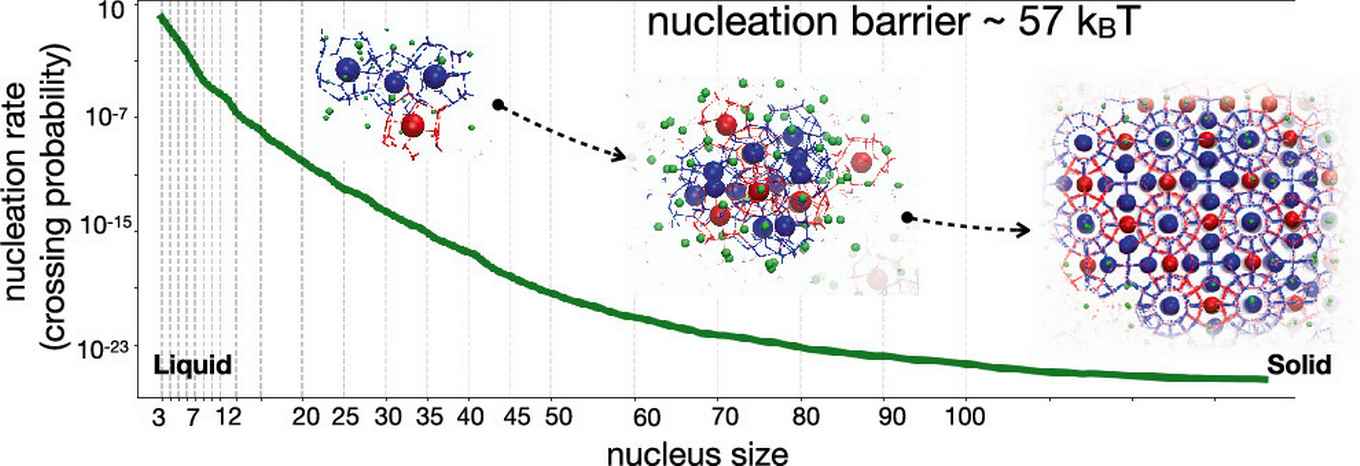
The crystallization of methane hydrates via homogeneous nucleation under natural, moderate conditions is of both industrial and scientific relevance, yet still poorly understood. Predicting the nucleation rates at such conditions is notoriously difficult due to high nucleation barriers, and requires, besides an accurate molecular model, enhanced sampling.
Crystal nucleation rate
Employing the efficient transition interface sampling technique, Arjun Wadhawan and Peter Bolhuis of the HIMS research group Computational Chemistry now predict the exact rate of nucleation with an accurate atomistic force field, focusing on specific conditions of 280 K and 500 bar. They computed a crystal nucleation rate of a few hundred nuclei per second per cm3. This figure is in agreement with experimental estimates for nearby conditions, although this is most likely fortuitous as the predictions are very sensitive to the precise simulation setup. Nevertheless, the work shows that it is now possible to compute rates for methane hydrates at moderate supersaturation, without relying on any assumptions other than the force field. This will assist future research aiming at understanding natural hydrates, improving materials synthesis, and developing dissolving strategies.
Paper
A. Arjun and P. G. Bolhuis: Rate Prediction for Homogeneous Nucleation of Methane Hydrate at Moderate Supersaturation Using Transition Interface Sampling. J. Phys. Chem. B 2020, 124, 37, 8099–8109 DOI: 10.1021/acs.jpcb.0c04582