Simple one-pot synthesis of druggable tricyclic peptides
New promising class of therapeutics could replace vancomycin and combat MRSA
18 December 2017
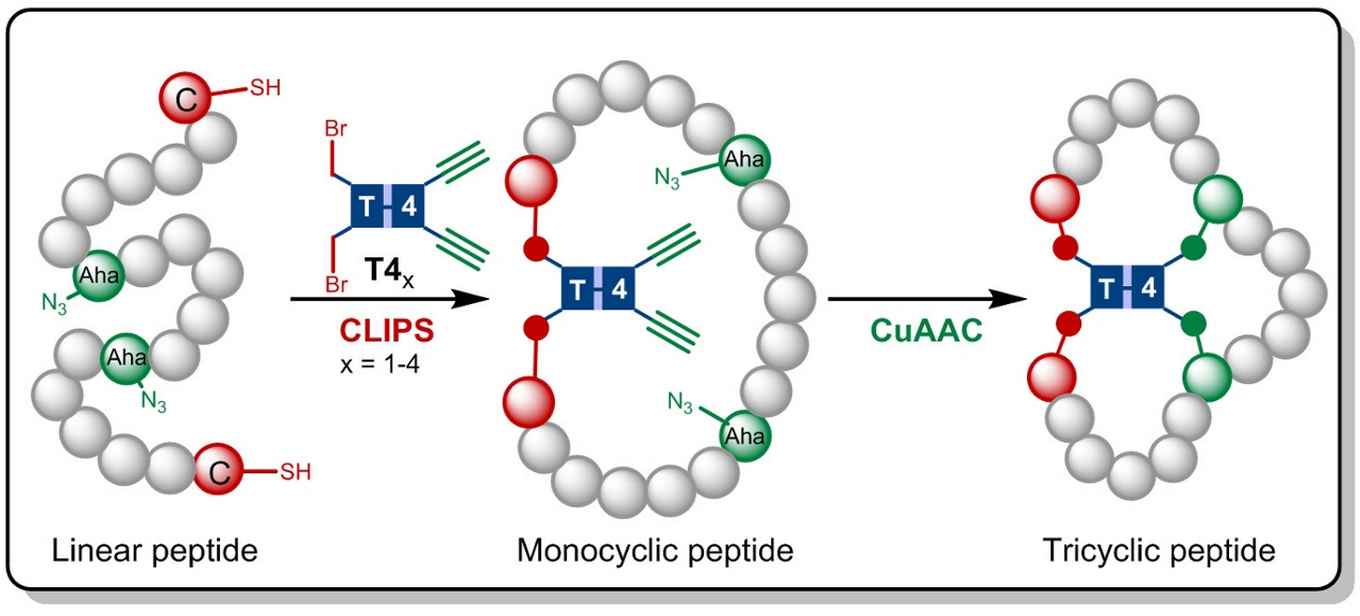
The methodology, developed by HIMS PhD student Gaston Richelle, enables the parallel synthesis of multiple tricyclic peptides in a library format. This permits subsequent biological screening in order to identify novel peptide therapeutics.
Higher level of structural complexity
Over the last decade cyclic peptides have emerged as a promising class of therapeutics, showing a wide therapeutic window that ranges from antifertility to antiviral and anticancer applications. Many monocyclic and bicyclic peptides have been identified and the number of cyclic peptides entering clinical trials has drastically increased.
Even so, it has become clear that in some cases more structurally complex peptides are needed to reach appropriate activity levels. Vancomycin, the 'last-resort' tricyclic antibiotic peptide, provides an illustrative example of such a complex construct. The alarmingly increasing resistance of bacteria against currently available antibiotics calls for complex peptide constructs in analogy to vancomycin.
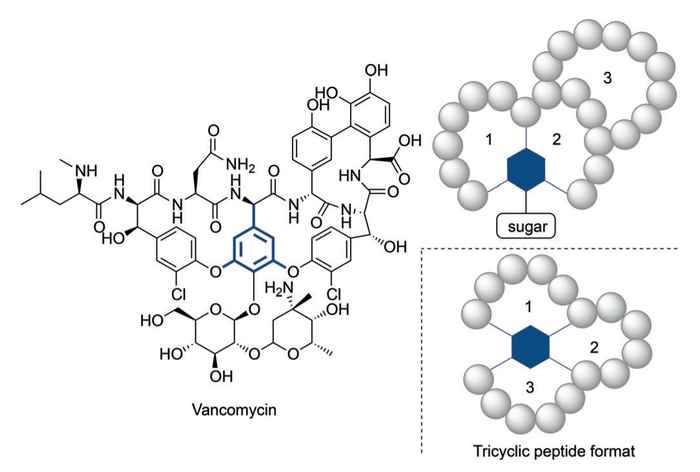
The search for novel synthetic routes to complex multicyclic peptides has thus gained serious interest. Previously reported methodologies for creating multicyclic peptides has been limited due to multiple isomer formation, laborious reaction protocols or limited structural diversity.
CLIPS combined with CuAAC
In the research now published in Angewandte Chemie the Amsterdam chemists build on the CLIPS technology earlier developed at Pepscan. This method for 'Chemical LInkage of Peptides onto Scaffolds' yields monocyclic- and bicyclic peptides that have shown to exhibit enhanced selectivities and affinities against target proteins.
Gaston Richelle has now combined the CLIPS method with the fully compatible CuAAC 'click' chemistry, leading to the formation of the complex tricyclic peptides. By making use of flexible scaffold molecules, the sought-after tricyclic peptides are formed in an isomerically pure fashion. The main advantage of the new methodology is that the CLIPS- and CuAAC-reactions can be carried out in a one-pot procedure with no limitations to the nature and amounts of amino acids that are implemented in the peptide loops.
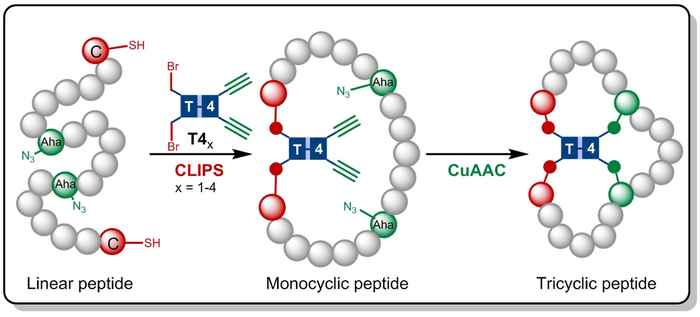
As a result the methodology can be applied in high-diversity peptide libraries to generate a broad range of tricyclic topologies that can be screened for biological activity. Currently multiple inhibition screenings are carried out, e.g. against enzymes that are highly abundant in first-stage cancer cells. Also novel antimicrobial activity studies are carried out against MRSA and Staphylococcus aureus. Adding to this, the research team is currently studying the use of even more complex T6 scaffolds to create penta- and hexacyclic peptides.
Article
Richelle, G. J. J., Ori, S., Hiemstra, H., van Maarseveen, J. H. and Timmerman, P. (2017), General and Facile Route to Isomerically Pure Tricyclic Peptides Based on Templated Tandem CLIPS/CuAAC Cyclizations. Angew. Chem. Int. Ed.. doi:10.1002/anie.201709127