Metrical oxidation states of 1,4-diazadiene derived ligands
16 February 2021
Abstract
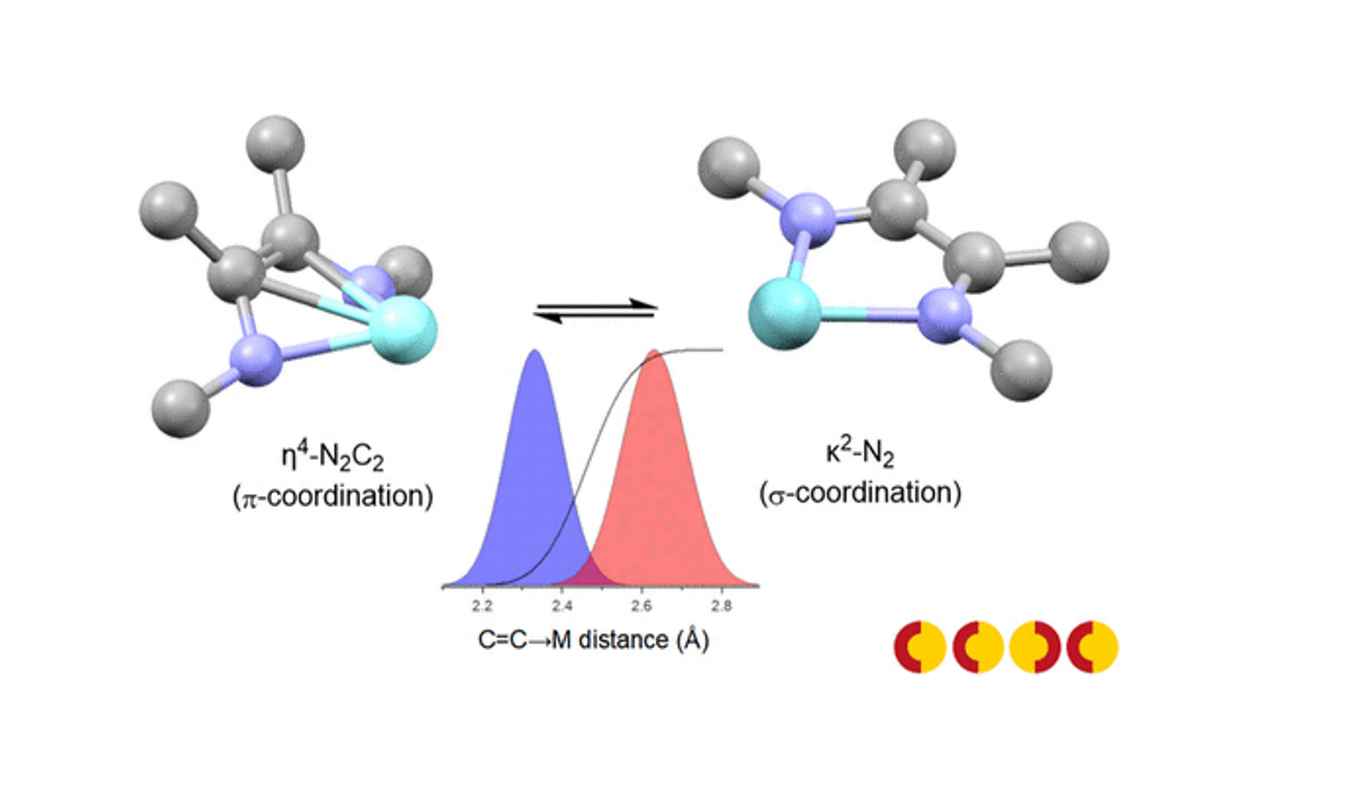
The conventional method of assigning formal oxidation states (FOS) to metals and ligands is an important tool for understanding and predicting chemical reactivity, in particular in catalysis research. For complexes containing redox-noninnocent ligands, the oxidation state of the ligand can be ambiguous (i.e., their spectroscopic oxidation state can differ from the formal oxidation state), and thus frustrates the assignment of the oxidation state of the metal. A quantitative correlation between empirical metric data of redox active ligands and their oxidation states using a metrical oxidation state (MOS) model has been developed for catecholate and aminophenolate derived ligands by Brown. In the present work, we present a MOS model for 1,4-diazabutadiene (DADn) ligands. The model is based on a similar approach as reported by Brown, correlating the intra-ligand bond lengths of the DADn moiety in a quantitative manner to the MOS using geometrical information from X-ray structures in the Cambridge Crystallographic Data Center (CCDC) database. However, accurate determination of the MOS of these ligands turned out to be dependent the coordination mode of the DAD2‒ moiety, which can adopt both a planar κ2-N2-geometry and a η4-N2C2 π-coordination mode in (transition) metal complexes in its doubly reduced, dianionic enediamide oxidation state. A reliable MOS model was developed taking the intrinsic differences in intra-ligand bond distances between these coordination modes of the DAD2‒ ligand into account. Three different models were defined and tested using different geometric parameters (C=C→M distance, M−N−C angle, M−N−C−C torsion angle) to describe the C=C backbone coordination to the metal in the η4-N2-C2 π-coordination mode of the DAD2‒ ligand. Statistical analysis revealed that the C=C→M distance best describes the η4-N2-C2 coordination mode, using a cut-off value of 2.46 Å for π-coordination. The developed MOS model was used to validate the oxidation state assignment of elements not contained within the training set (Sr, Yb and Ho), thus demonstrating the applicability of the MOS model to a wide range of complexes. Chromium complexes with complex electronic structures were also shown to be accurately described by MOS analysis. Furthermore, it is shown that a combination of MOS analysis and FOD calculations provide an inexpensive method to gain insight into the electronic structure of singlet spin state (S = 0) [M(trop2dad)] transition metal complexes showing (potential) singlet biradical character.
Paper
Felix J. de Zwart, Bente Reus, Annechien A.H. Laporte, Vivek Sinha and Bas de Bruin: Metrical oxidation states of 1,4-diazadiene (DAD) derived ligands. Inorganic Chemistry, publication date February 15, 2021. DOI: 10.1021/acs.inorgchem.0c03685 (preprint repository: 10.26434/chemrxiv.13378967.v1)
Link
Website HomKat research group: Heterogeneous, Supramolecular and Bio-inspired Chemistry