Unraveling reaction mechanisms via machine learning
21 September 2021
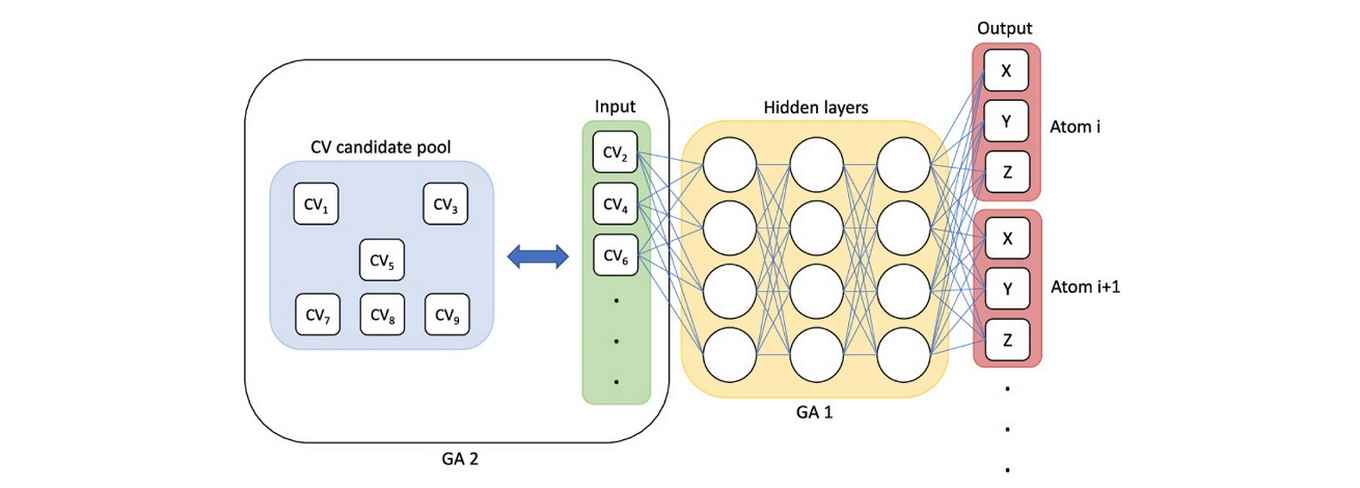
The FABULOUS software, developed by Ferry Hooft and Alberto Pérez de Alba Ortíz, has been made freely available on GitHub. The work was presented at the recent Lorentz Center Workshop on “Accelerating the Understanding of Rare Events” in Leiden. It has been published in a paper in the Journal of Chemical Theory and Computation:
Abstract
With the continual improvement of computing hardware and algorithms, simulations have become a powerful tool for understanding all sorts of (bio)molecular processes. To handle the large simulation data sets and to accelerate slow, activated transitions, a condensed set of descriptors, or collective variables (CVs), is needed to discern the relevant dynamics that describes the molecular process of interest. However, proposing an adequate set of CVs that can capture the intrinsic reaction coordinate of the molecular transition is often extremely difficult. Here, we present a framework to find an optimal set of CVs from a pool of candidates using a combination of artificial neural networks and genetic algorithms. The approach effectively replaces the encoder of an autoencoder network with genes to represent the latent space, i.e., the CVs. Given a selection of CVs as input, the network is trained to recover the atom coordinates underlying the CV values at points along the transition. The network performance is used as an estimator of the fitness of the input CVs. Two genetic algorithms optimize the CV selection and the neural network architecture. The successful retrieval of optimal CVs by this framework is illustrated at the hand of two case studies: the well-known conformational change in the alanine dipeptide molecule and the more intricate transition of a base pair in B-DNA from the classic Watson–Crick pairing to the alternative Hoogsteen pairing. Key advantages of our framework include the following: optimal interpretable CVs, avoiding costly calculation of committor or time-correlation functions, and automatic hyperparameter optimization. In addition, we show that applying a time-delay between the network input and output allows for enhanced selection of slow variables. Moreover, the network can also be used to generate molecular configurations of unexplored microstates, for example, for augmentation of the simulation data.
Publication details
Ferry Hooft, Alberto Pérez de Alba Ortíz, and Bernd Ensing: Discovering Collective Variables of Molecular Transitions via Genetic Algorithms and Neural Networks. J. Chem. Theory Comput. 17, 4, 2294–2306 (2021) DOI: 10.1021/acs.jctc.0c00981