Efficient computational optimisation of catalysis kinetics
12 August 2025
Catalysis plays an important role in many fields, from industrial chemistry to the molecular biology of the cell. The efficiency of a catalyst for accelerating a reaction strongly depends on its molecular structure and interactions with the substrate. In principle, given a particular molecular interaction model, one could predict kinetics of a catalytic cycle and, by extension, provide optimal interaction parameters.
However, this is usually considered computationally prohibitively expensive, in particular for solvated catalytic systems such as enzymes and DNA-decorated colloidal particles. While for gas phase reactions quantum chemistry provides accurate predictions, condensed-phase (solvated) reactions, and in general reactions in high-dimensional systems, require a dynamical approach. This renders computation exceptionally expensive, even when considering simplified classical molecular models.
Improving the catalytic turnover by orders of magnitude
In the PNAS paper, Bolhuis describes how an inverse design approach (see figure) employing a novel path reweighting method offers a way out of this computational conundrum.
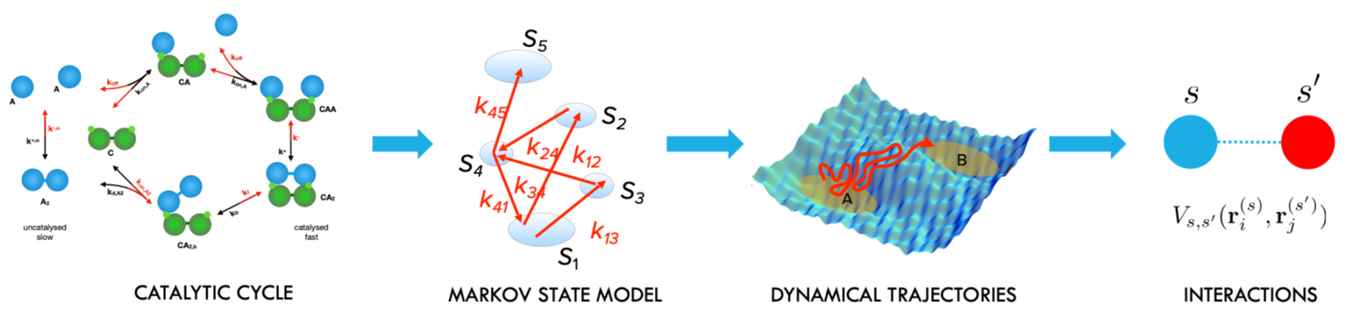
The approach features computing the ensemble of trajectories using molecular dynamics for a single parameter setting of the molecular interactions and then expanding the kinetic landscape around these parameters. As a result, it becomes possible to improve the optimal catalytic turnover by orders of magnitude in one single simulation.
Efficient computational design
The versatility of the methodology was showcased on minimal models for dimerization and kinase signalling, highlighting its potential for efficient computational design of complex catalysts using more realistic models. Since the methodology is capable of identifying the effect of each model parameter, the approach can be used to improve on such molecular models, e. g. by suggesting mutations or substitution of functional groups. For instance, in the kinase model the location of the binding site might be altered through protein engineering. Bolhuis envisions that path reweighting techniques will become part of the molecular simulation toolbox in the near future.
Paper details
Peter G. Bolhuis: Optimal kinetics for catalytic cycles from a single path-sampling simulation. Proc. Natl. Acad. Sci. U.S.A. 122 (30) e2500934122 (2025). DOI: 10.1073/pnas.2500934122