Understanding mechanisms of molecular rare events from start to finish
Efficient computer simulation maps full molecular reaction mechanism
24 April 2026
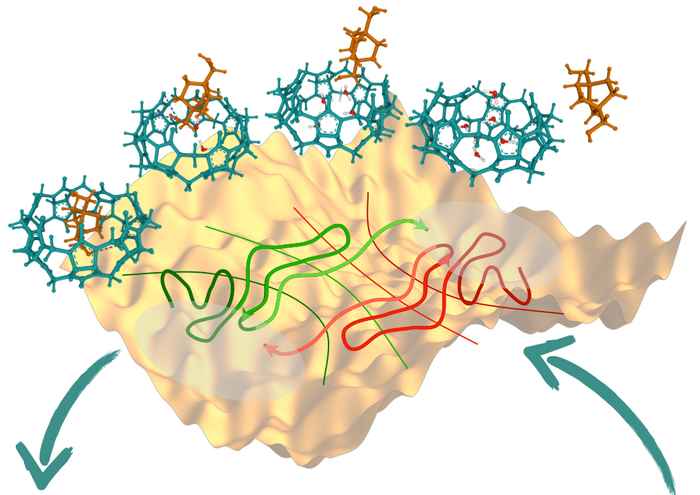
A chemical reaction is usually initiated by reactant molecules overcoming an energy barrier, leading to the rearrangement of atoms and/or electrons between the molecules, ultimately yielding a final product. Although unlikely, such rare reactive events are all-determining for the actual chemical process. The reaction mechanism describes in molecular detail how the overall process occurs from the initial to the final state. It can be quite complex, involving several sub-steps, or even multiple parallel reaction channels.
Experimentally elucidating such mechanisms in detail is quite a challenge, as they usually occur on extremely fast timescales. Computer simulations and modelling can help here. Over the years, many approaches have been developed to computationally study such rare event dynamics. Mathematically, the reaction mechanism can be captured in a so-called committor function, which represents for each point in the process the probability of reaching the final state. As such, it provides a means to predict and understand the reaction.
Computing the full reaction sequence
However, mapping the committor function for the entire reactive process has for a long time been out of reach because of the sheer computational effort needed to achieve this. In their study presented in Physical Review Letters, PhD student Rik Breebaart, together with his supervisor Prof. Peter Bolhuis and collaborators from Goethe University Frankfurt, have now overcome this challenge. They have for the first time managed to map the committor function for a full reaction sequence, capturing the process in complete detail from beginning to end. The researchers used a neural network and implemented an iterative, active learning scheme based on trajectory sampling. The resulting simulation was capable of predicting probabilities to an unprecedented degree of 1 in 10-20, which amounts to finding one reactive trajectory in a billion times a billion unreactive trajectories.
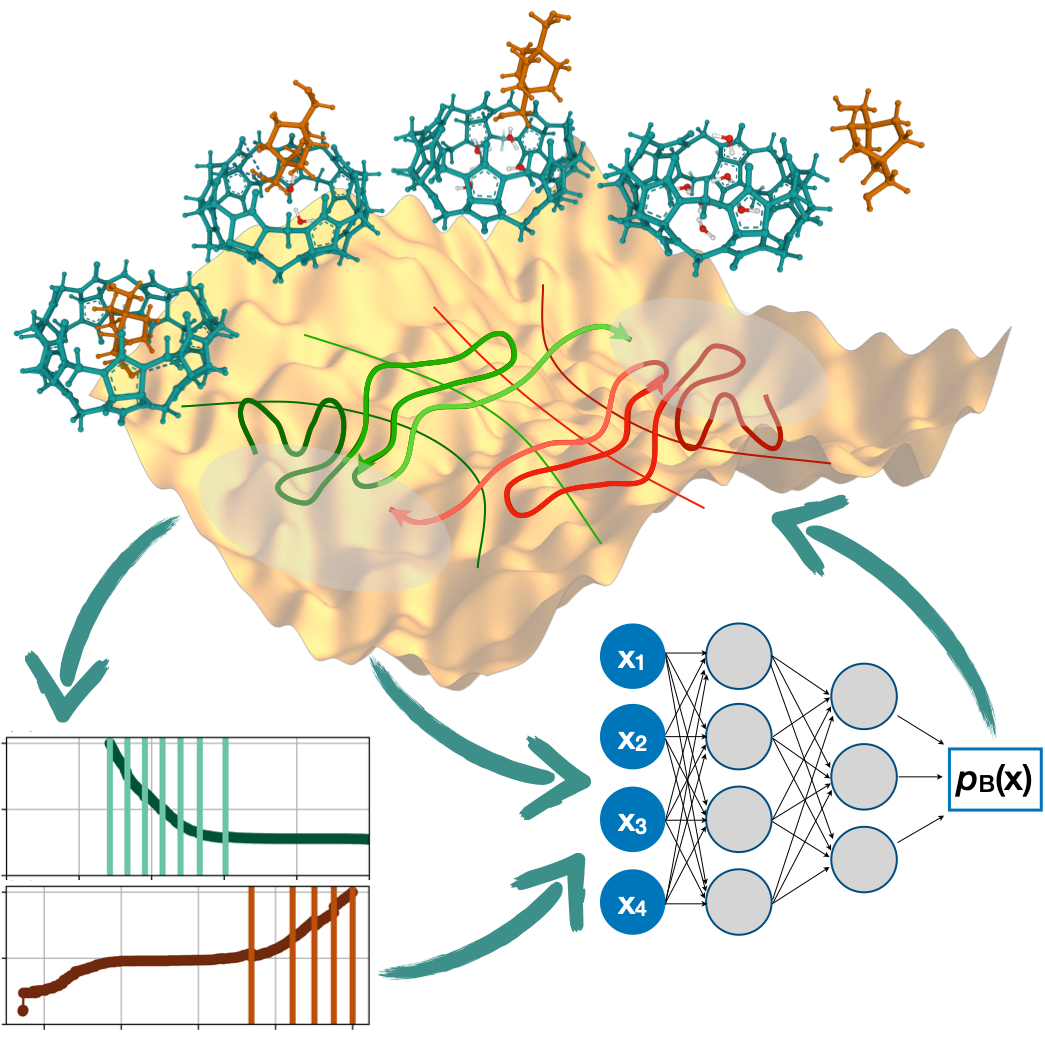
The paper describes the simulation of a ligand binding reaction where a specific molecule is being attached to a protein. The methodology yielded accurate predictions of the reaction rate, revealed the presence of metastable states, and helped identify the most important ingredients in the process as a function of the committor. This revealed unexpected features of the reaction, such as the number of water molecules in the binding pocket of the protein, and the orientation of the ligand necessary for binding/unbinding along the entire process.
Valuable insights and control opportunities
The mechanistic understanding that can be achieved with the novel simulation approach not only yields valuable insights into the underlying physical processes. It also offers control strategies, reveals key reaction elements, and informs future experimental design. The results presented in the paper show that the methodology opens up many possibilities to discover novel and poorly understood mechanisms in complex systems.
Abstract of the paper
Understanding mechanisms of rare but important events in complex molecular systems, such as protein folding or ligand (un)binding, requires accurately mapping transition paths from an initial to a final state. The committor is the ideal reaction coordinate for this purpose, but calculating it for high-dimensional, nonlinear systems has long been considered intractable. Here, we introduce an iterative path sampling strategy for computing the committor function for systems with high free energy barriers. We start with an initial guess to define isocommittor interfaces for transition interface sampling. The resulting path ensemble is then reweighted and used to train a neural network, yielding a more accurate committor model. This process is repeated until convergence, effectively solving the long-standing circular problem in enhanced sampling where a good reaction coordinate is needed to generate efficient sampling, and vice-versa. The final, converged committor model can be interrogated to extract mechanistic insights. We demonstrate the power of the method on a benchmark 2D potential and a host-guest (un)binding process in explicit solvent.
Paper details
R.S. Breebaart, G. Lazzeri, R. Covino and P.G. Bolhuis, Understanding mechanisms of molecular rare events from start to finish. Phys. Rev. Lett, 2026. DOI: 10.1103/lk32-njx7