Calibrating a computational microscope
4 January 2021
Elucidation of biomolecular structures
When studying biomolecules such as proteins or DNA, a first step often consists in the determination of their structures, for example, using X-ray crystallography or cryo-electron microscopy. By revealing a wide range of structure-function relationships, this approach has enabled major advances in molecular biology in the last 50 years. Such experiments are often combined with computational methods, which yield the motions of all the atoms in a molecular system as dictated by thermal fluctuations.
These molecular simulations provide a computational microscope that can resolve atomistic structures, otherwise elusive in experiments. However, the description of the interactions between atoms is often approximate, which can lead to results that are not entirely accurate. Statistical methods developed in the last decade are now able to increase the accuracy of molecular dynamics simulations by incorporating experimental information, and allow the determination of thermodynamic ensembles of structures.
However, one would like to go even further, by obtaining kinetic ensembles, comprising the structures of the different states of a molecular system, their populations and their interconversion rates. The latter are even harder to predict correctly. To fine tune the computational microscope one long-standing idea was to make use of knowledge about experimental rates. However, up to now it was still unclear how to include this experimental knowledge into the simulations.
Unleashing the power of the maximum-caliber principle
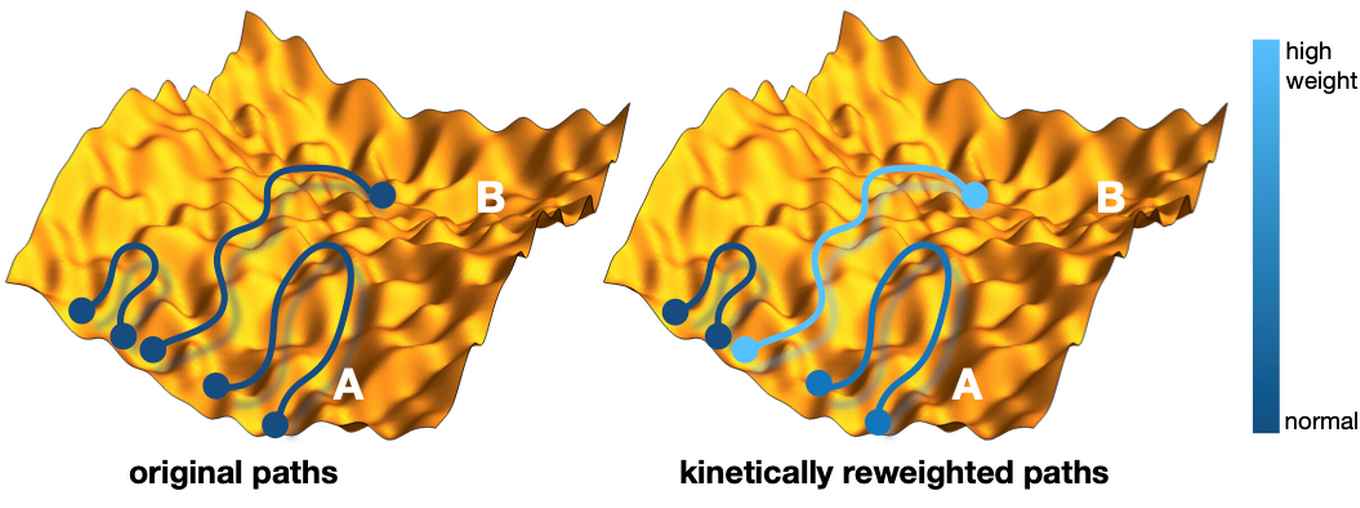
The aim of the Amsterdam and Cambridge team was to make a first step in this direction by introducing a method of imposing known rate constants as constraints in molecular dynamics simulations, which is based on a combination of the maximum-entropy (MaxEnt) and maximum-caliber principles (MaxCal). Originating from information theory, the MaxEnt and MaxCal principles are powerful concepts in statistical physics and yield optimal models given imposed constraints. Starting from an existing ensemble of trajectories, the novel method reweights each path in order to match the calculated and experimental interconversion rates of a molecular transition of interest, while minimally perturbing the prior path distribution (see figure for a schematic representation of this reweighting). This kinetically corrected ensemble of trajectories leads to improved structure, kinetics and thermodynamics, as well mechanistic insights that may not be readily evident directly from the experiments.
Protein folding
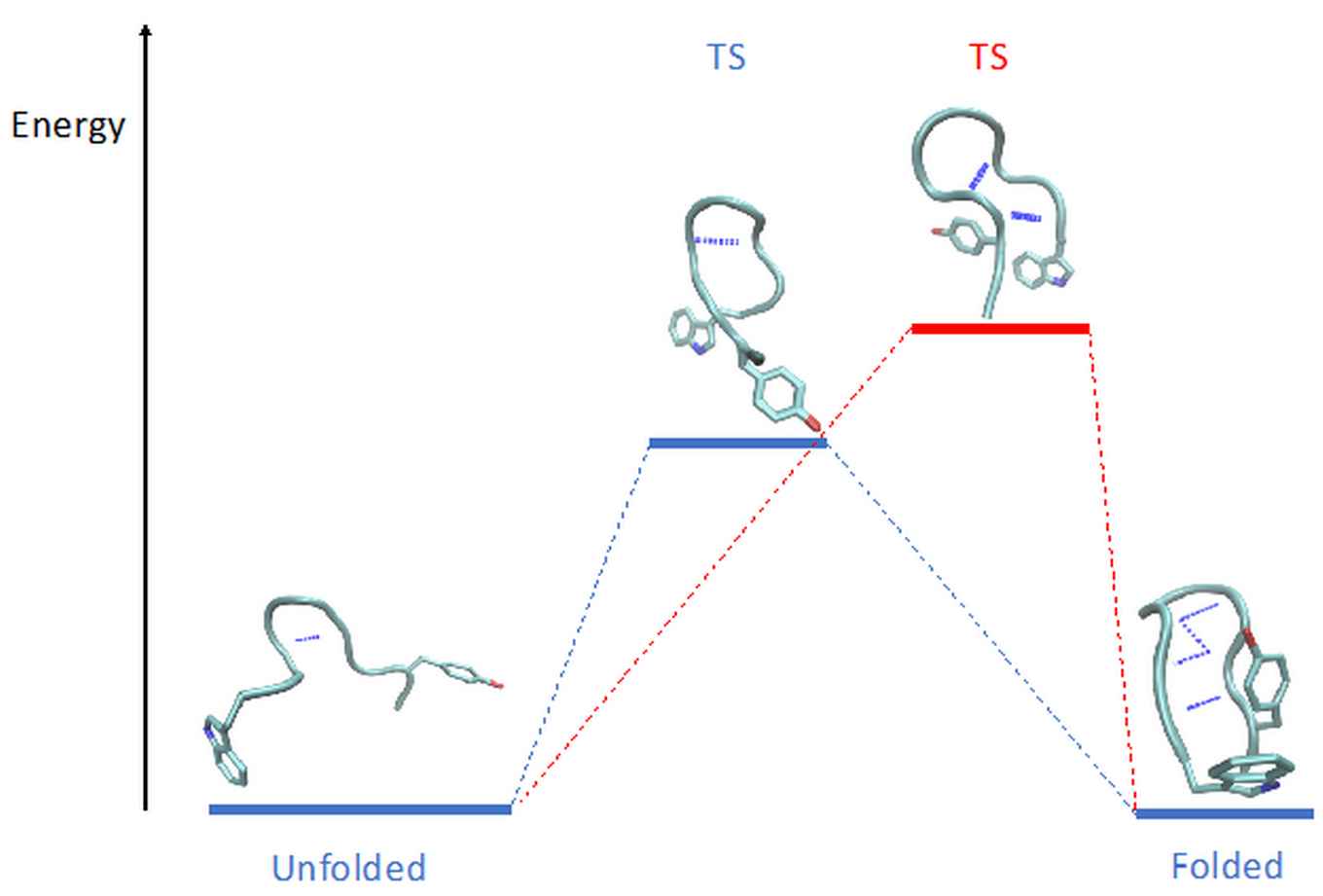
Brotzakis et al. demonstrated their method on the determination of transition states, reaction mechanisms and free energies of all atom molecular simulations of protein folding, which are notoriously difficult. Protein folding is a crucial process in biology, and while we have reached a better understanding over the past decades, the accurate prediction of the kinetic mechanism of protein folding is still elusive. As a starting point the team borrowed prior molecular dynamics simulations previously run on Anton, the fastest molecular dynamics computer in existence. Due to inevitable approximations in the simulations, the rate predictions were off compared to experiment. The new method managed to correct for this mismatch, and found a shift in the transition state, the halfway point between an unfolded and folded state, to a structure closer to final fold (see figure).
The researchers anticipate that this method will extend the applicability of molecular simulations to kinetic studies in structural biology, and that it will assist the development of force fields to reproduce kinetic and thermodynamic observables. Michele Vendruscolo, at Cambridge, part of the team, concludes “As the outcomes of molecular processes often depend on the rates at which they occur, with this new method we can improve our ability to determine molecular mechanisms responsible for biomolecular function and dysfunction”. Peter Bolhuis of HIMS adds enthusiastically “Correcting the kinetic flaw in the computational microscope allows us to suddenly see much sharper. Moreover, we think this approach is generally applicable to a wide range of systems in biology, physics, chemistry, and material science. As such, it opens up many possibilities for improving our understanding of nature at the microscopic scales”.
Publication
Brotzakis, Z. F., Vendruscolo, M., & Bolhuis, P. G. (2021). A method of incorporating rate constants as kinetic constraints in molecular dynamics simulations. Proceedings of the National Academy of Sciences, 118(2), e2012423118. https://doi.org/10.1073/pnas.2012423118
Preprint repository
https://arxiv.org/abs/2006.00868