Machine learning for prediction of peak width in HRMS data
Interconverting data from centroided to profile mode
14 December 2021
Abstract
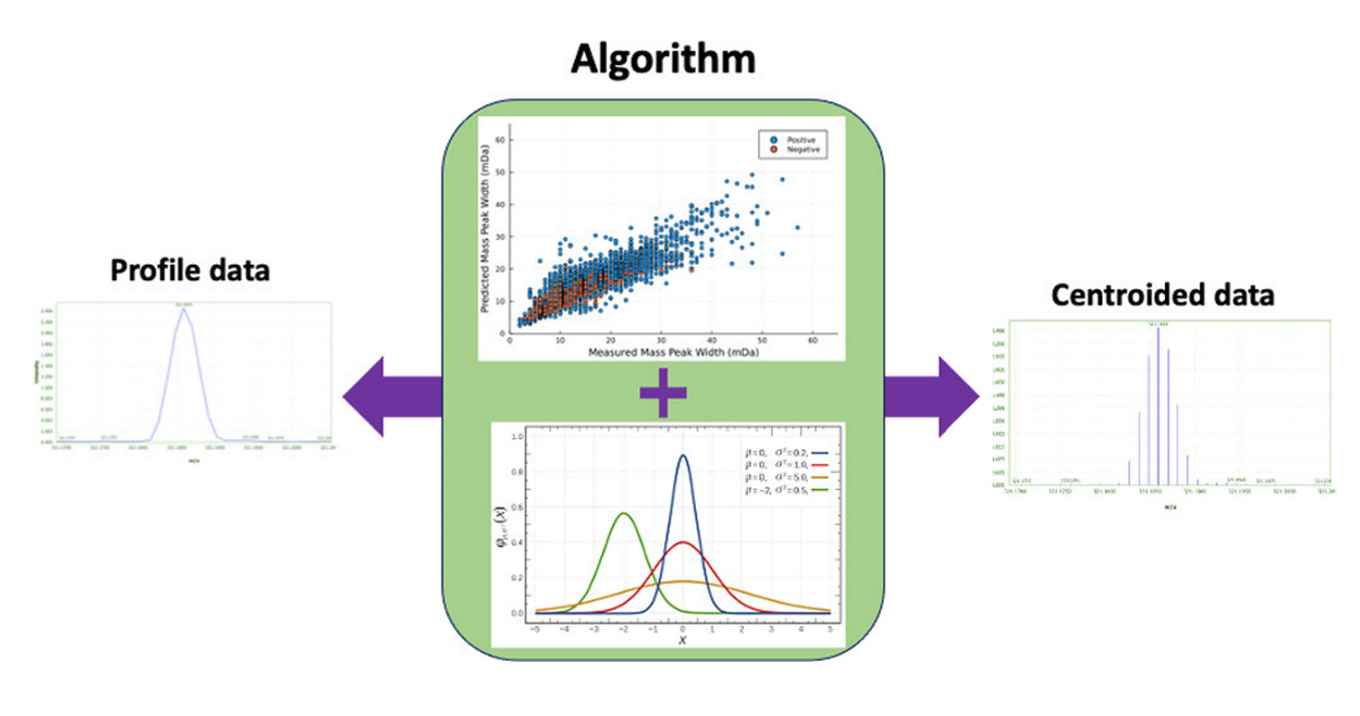
Centroiding is one of the major approaches used for size reduction of the data generated by high-resolution mass spectrometry. During centroiding, performed either during acquisition or as a pre-processing step, the mass profiles are represented by a single value (i.e., the centroid). While being effective in reducing the data size, centroiding also reduces the level of information density present in the mass peak profile. Moreover, each step of the centroiding process and their consequences on the final results may not be completely clear. Here, we present Cent2Prof, a package containing two algorithms that enables the conversion of the centroided data to mass peak profile data and vice versa. The centroiding algorithm uses the resolution-based mass peak width parameter as the first guess and self-adjusts to fit the data. In addition to the m/z values, the centroiding algorithm also generates the measured mass peak widths at half-height, which can be used during the feature detection and identification. The mass peak profile prediction algorithm employs a random-forest model for the prediction of mass peak widths, which is consequently used for mass profile reconstruction. The centroiding results were compared to the outputs of the MZmine-implemented centroiding algorithm. Our algorithm resulted in rates of false detection ≤5% while the MZmine algorithm resulted in 30% rate of false positive and 3% rate of false negative. The error in profile prediction was ≤56% independent of the mass, ionization mode, and intensity, which was 6 times more accurate than the resolution-based estimated values.
Publication details
Saer Samanipour, Phil Choi, Jake W. O’Brien, Bob W. J. Pirok, Malcolm J. Reid, and Kevin V. Thomas: From Centroided to Profile Mode: Machine Learning for Prediction of Peak Width in HRMS Data. Anal. Chem, November 29, 2021, DOI: 10.1021/acs.analchem.1c03755