Synthetic relevance of unique cobalt(III) N-enolate carbene radicals
24 March 2022
Cyclopropanes are important substructures in specific medicines and several other bioactive molecules. However, their synthetic applications are limited in particular because the synthesis of disubstituted acceptor-acceptor cyclopropanes under mild conditions using base-metal catalysts has hardly been established. This is in part due to the fact that the diazo precursors that do allow the generation of carbenes with two acceptor substituents are typically difficult to activate by commonly applied base-metal catalysts.
The Amsterdam researchers took a different approach and investigated the use of iodonium ylides instead of diazo precursors. Iodonium ylides are more powerful carbene delivering precursors, but precisely because of their enhanced reactivity they are also well-known to trigger 'over-carbenation' of the catalyst. With too many carbene units generated at the catalyst, ligand modification and active site blocking is often observed with catalyst deactivation typically being the end result.
Beneficial N-enolate formation
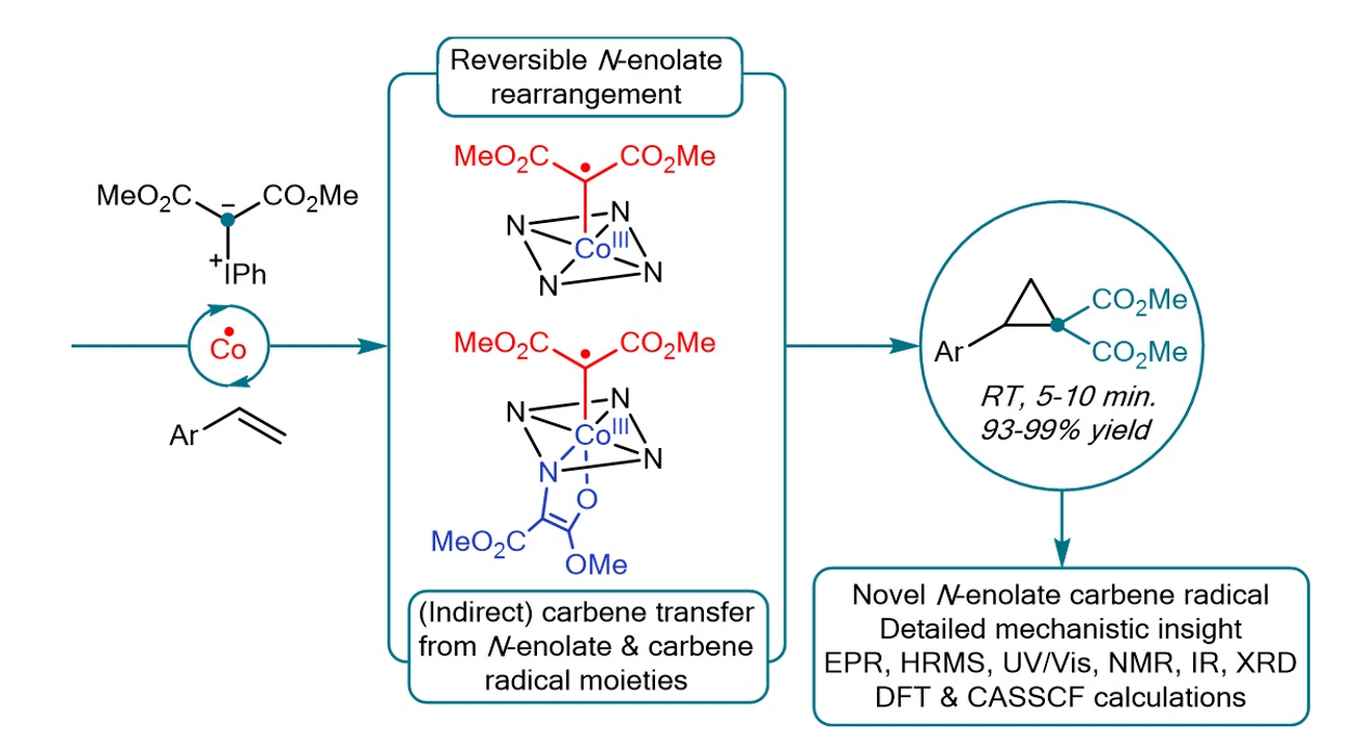
The Amsterdam researchers investigated cobalt(II)-catalyzed cyclopropanation reactions using iodonium ylides as disubstituted acceptor-acceptor carbene precursors. This system holds the risk of formation of N-enolates blocking catalyst activity as a result of bridging between the metal and a pyrrole unit of the porphyrin ligand. By performing a thorough mechanistic analysis, the researchers now show that this N-enolate formation strongly depends on the electronic structure of the catalyst. In particular, they demonstrate that for paramagnetic metalloradical cobalt(II) porphyrin complexes N-enolate formation is in fact beneficial. Due to the unique electronic structure of these catalysts, N‑enolates are in equilibrium with carbene radical intermediates, and hence their formation is reversible. The N-enolate even has a protective function, slowing down catalyst deactivation via hydrogen atom transfer (HAT) from the solvent. The study thus reveals a unique interplay between the reactivity of hypervalent iodonium ylides and the open-shell metalloradical cobalt(II)-tetraphenylporphyrin complex in catalytic cyclopropanation reactions, enabling fast and high-yielding reactions at room temperature. Future application of these unique species holds the potential to unlock new scaffolds via the superior activation of acceptor-acceptor iodonium ylides over diazo compounds.
Abstract
Square-planar cobalt(II)-systems have emerged as powerful carbene transfer catalysts for the synthesis of numerous (hetero)cyclic compounds via cobalt(III)-carbene radical intermediates. Spectroscopic detection and characterization of reactive carbene radical intermediates is limited to a few scattered experiments, centering around mono-substituted carbenes. Here, we reveal the unique formation of disubstituted cobalt(III)-carbene radicals derived from a cobalt(II)-tetraphenylporphyrin complex and acceptor–acceptor iodonium ylides as carbene precursors and their catalytic application. Particularly noteworthy is the fact that iodonium ylides generate novel bis-carbenoid species via reversible ligand modification of the paramagnetic [Co(TPP)]-catalyst. Two interconnected catalytic cycles are involved in the overall mechanism, with a mono-carbene radical and an unprecedented N-enolate-carbene radical intermediate at the heart of each respective cycle. Notably, N-enolate formation is not a deactivation pathway but a reversible process, enabling transfer of two carbene moieties from a single N-enolate-carbene radical intermediate. The findings are supported by extensive experimental and computational studies.
Publication details
Roel F. J. Epping, Mees M. Hoeksma, Eduard O. Bobylev, Simon Mathew, Bas de Bruin: Cobalt(II)–tetraphenylporphyrin-catalysed carbene transfer from acceptor–acceptor iodonium ylides via N-enolate–carbene radicals. Nature Chemistry, published online 21 March 2022. DOI: 10.1038/s41557-022-00905-4
Links
- Research project of Roel Epping
- Research group of Bas de Bruin
- Metalloradical catalysis at the Homogeneous, Supramolecular and Bio-Inspired Catalysis research group