Using machine learning to analyse samples with many unknown chemical components
28 March 2023
Chemicals are everywhere: our environment and our body contain countless chemicals of all sorts. They come in a wide variety of molecular weights, functionalities, physiochemical properties, and toxicity. Analytical scientist develop methods to provide insight in the mix of chemicals we are exposed to, dubbed the environmental and human ‘exposome’. However, estimating the potential health impact of this exposure is difficult because many of the chemicals are structurally unknown. The latter complicates the interpretation of results when applying the widely used analytical strategy of liquid chromatography followed by mass spectrometry (LC-MS). A key parameter in this analysis is the retention index (Ri), of individual components, which can only be estimated based on their chemical structure.
In the paper in the Journal of Cheminformatics, PhD students Jim Boelrijk (IvI) and Denice van Herwerden (HIMS) now present a novel machine learning algorithm that can predict the retention indices of chemicals for structurally unknown species in non-target analysis combined with high-resolution mass spectrometry. They performed their research in cooperation with researchers Bernd Ensing (HIMS) and Patrick Forré (IvI) at the joint AI4Science Laboratory, and Sear Samanipour (HIMS) at the UvA Data Science Centre. They validated their approach using both experimental Ri values and descriptor-based predicted Ri values, and showed comparable accuracy to conventional molecular descriptor-based models.
Abstract
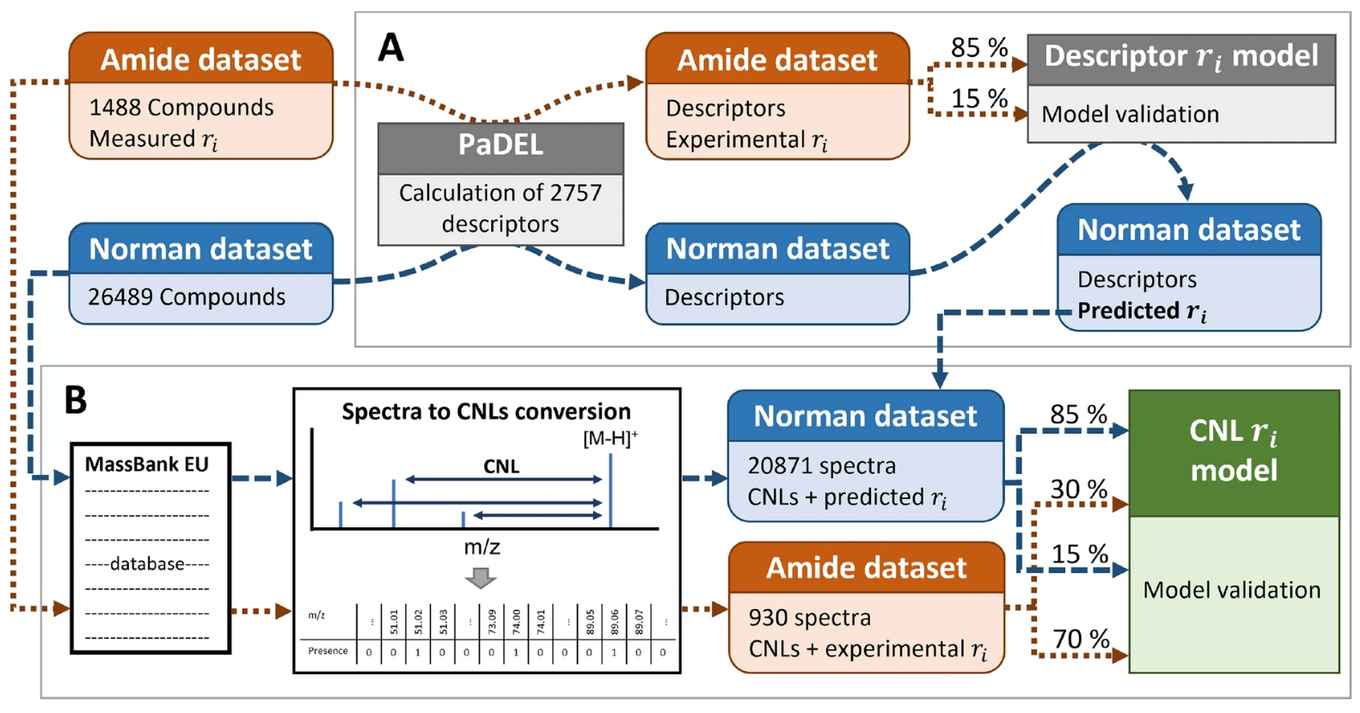
Non-target analysis combined with liquid chromatography high resolution mass spectrometry is considered one of the most comprehensive strategies for the detection and identification of known and unknown chemicals in complex samples. However, many compounds remain unidentified due to data complexity and limited number structures in chemical databases. In this work, we have developed and validated a novel machine learning algorithm to predict the retention index (ri) values for structurally (un)known chemicals based on their measured fragmentation pattern. The developed model, for the first time, enabled the predication of ri values without the need for the exact structure of the chemicals, with an R2 of 0.91 and 0.77 and root mean squared error (RMSE) of 47 and 67 ri units for the NORMAN (n=3131) and amide (n=604) test sets, respectively. This fragment based model showed comparable accuracy in ri prediction compared to conventional descriptor-based models that rely on known chemical structure, which obtained an R2 of 0.85 with an RMSE of 67.
The algorithms for ri prediction, including the trained models and leverage matrices are available at GitHub. In addition, a Google Collab tutorial is available on usability of the models and the use of datasets.
Publication details
Jim Boelrijk, Denice van Herwerden, Bernd Ensing, Patrick Forré & Saer Samanipour: Predicting RP-LC retention indices of structurally unknown chemicals from mass spectrometry data. J Cheminform 15, 28 (2023). DOI: 10.1186/s13321-023-00699-8