Binding dynamics of a stapled peptide targeting transcription factor NF-Y
11 April 2024
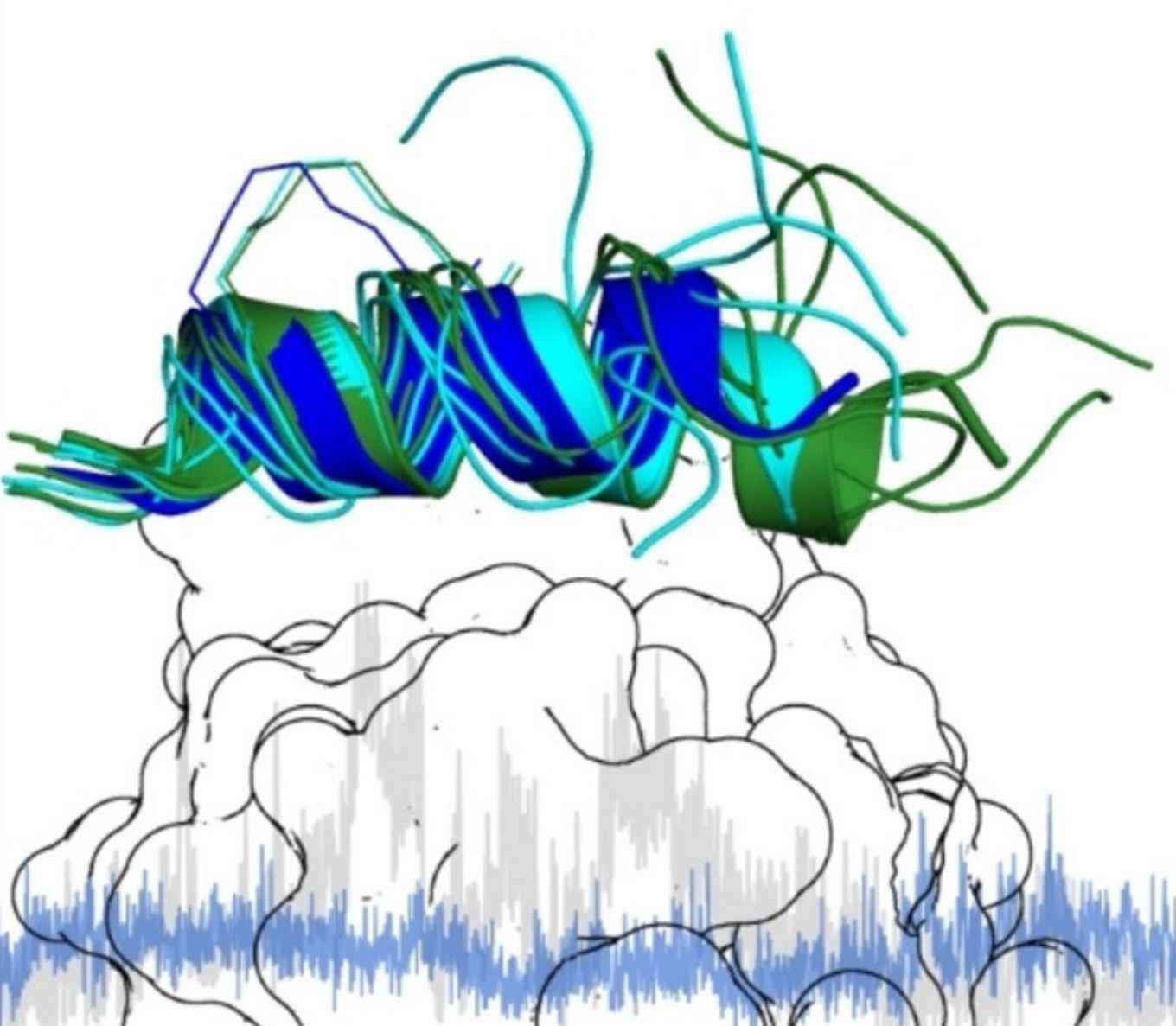
Due to the persistent challenges in targeting transcription factors, there is a demand for tool compounds that can effectively address this challenging class of proteins. Among these compounds are peptides and peptidomimetics that can be chemically modified for enhanced biological functions, including increased target affinity and serum stability. The study now published in ChemBioChem aimed to understand the impact of miniaturization on the crucial core binding region of a peptidomimetic binder targeting the NF−Y transcription factor.
Abstract of the paper
Transcription factors (TFs) play a central role in gene regulation, and their malfunction can result in a plethora of severe diseases. TFs are therefore interesting therapeutic targets, but their involvement in protein-protein interaction networks and the frequent lack of well-defined binding pockets render them challenging targets for classical small molecules. As an alternative, peptide-based scaffolds have proven useful, in particular with an α-helical active conformation. Peptide-based strategies often require extensive structural optimization efforts, which could benefit from a more detailed understanding of the dynamics in inhibitor/protein interactions. In this study, we investigate how truncated stapled α-helical peptides interact with the transcription factor Nuclear Factor-Y (NF-Y). We identified a 13-mer minimal binding core region, for which two crystal structures with an altered C-terminal peptide conformation when bound to NF-Y were obtained. Subsequent molecular dynamics simulations confirmed that the C-terminal part of the stapled peptide is indeed relatively flexible while still showing defined interactions with NF-Y. Our findings highlight the importance of flexibility in the bound state of peptides, which can contribute to overall binding affinity.
Paper details
C. Durukan, F. Arbore, R. Klintrot, C. Bigiotti, I.M. Ilie, J. Vreede, T.N. Grossmann, S. Hennig: Binding dynamics of a stapled peptide targeting the transcription factor NF‐Y, ChemBioChem 2024, e202400020. DOI: 10.1002/cbic.202400020
See also
Research group Computational Chemistry, Biomolecular simulations.