AI-improved prediction of thermodynamic properties of liquids
Machine-learned 'free energy functional' enhances efficiency and accuracy of thermodynamic calculation
13 February 2025
Although less famous than the quantum version used for electronic structure calculations, classical density functional theory (cDFT) remains a very appealing approach to describe fluids. However, similar as in quantum DFT, the accuracy of the theory is hampered by the lack of an exact description of the (free) energy functional. Over the years, various approximations have been developed, albeit with mixed success.
Now, a team of researchers at the Computational Chemistry Group (Van ‘t Hoff Institute for Molecular Sciences) and the AMLab (Informatics Institute) of the University of Amsterdam, together with the Debye Institute for Nanomaterials Science and the Institute for Theoretical Physics of Utrecht University present a machine-learned approximation that can significantly improve cDFT calculations of fluids and vapours.
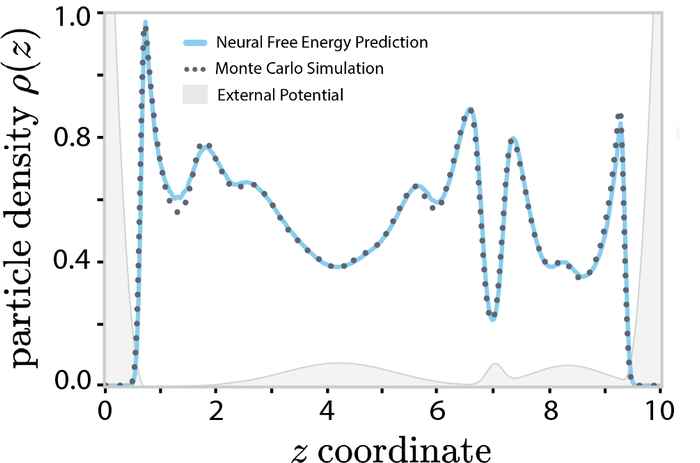
Their ‘neural density functional’ can be trained in two manners. The first approach applies the first functional derivative of the free energy, which requires for training a large dataset of inhomogeneous particle densities on a 3D grid computed with Monte Carlo simulations under various external potentials. This is computationally rather demanding. The second approach, based on the second functional derivatives, requires a dataset of only 1D radial distribution functions of only homogeneous densities. This requires significantly fewer simulations.
Remarkably, the authors were able to show that the second approach works as well as the first for a simple Lennard-Jones liquid in a 1D planar symmetry. Work is underway to apply the new method to more practical applications.
First author of the paper is PhD student Jacob Dijkman whose appointment is sponsored by the UvA Data Science Center; the DCS is gratefully acknowledged.
Abstract as published in the paper:
The intrinsic Helmholtz free-energy functional, the centerpiece of classical density functional theory, is at best only known approximately for 3D systems. Here we introduce a method for learning a neural-network approximation of this functional by exclusively training on a dataset of radial distribution functions, circumventing the need to sample costly heterogeneous density profiles in a wide variety of external potentials. For a supercritical Lennard-Jones system with planar symmetry, we demonstrate that the learned neural free-energy functional accurately predicts inhomogeneous density profiles under various complex external potentials obtained from simulations.
Paper details:
Jacobus Dijkman, Marjolein Dijkstra, René van Roij, Max Welling, Jan-Willem van de Meent, and Bernd Ensing: Learning neural free-energy functionals with pair-correlation matching. Phys. Rev. Lett. 134, 056103 (2025) DOI: 10.1103/PhysRevLett.134.056103
See also
• UvA HIMS Computational Chemistry Group
• AMLab | Amsterdam Machine Learning Lab
• Utrecht University, Soft Condensed Matter Group
• Utrecht University, Cond-Matter Theory